
Синдром Дауна у человека проявляется при трисомии по 21 паре хромосом. Объясните причины появления такого хромосомного набора у человека.
Спрятать пояснение
Пояснение.
1. При нарушении мейоза возникает нерасхождение хромосом у женщин.
2. Формируются аномальные клетки (XX) вместо нормальных гамет.
3. При оплодотворении гамета с аномальным набором 21-й пары хромосом (XX) сливается с нормальным сперматозоидом, содержащим в ядре одну хромосому 21-й пары. В результате формируется зигота с набором хромосом по 21-й паре — XXX.
Примечание к пункту 1.
Частота рождения больных детей коррелируется с возрастом родителей. Чем старше родители, тем выше риск рождения у них ребенка с лишней хромосомой. Так, риск иметь больного ребенка у женщины после 35 лет примерно в 10 раз выше, чем у 20-летней. Появление больного ребенка не зависит от того, каким по счету он рождается. Рождение ребенка с хромосомной патологией зависит и от возраста отца. Около 20% случаев трисомии обусловлено нерасхожденнем хромосом у отца, причем частота его у мужчин существенно возрастает после 45 лет.
Спрятать критерии
Критерии проверки:
| Критерии оценивания выполнения задания | Баллы |
|---|---|
| Ответ включает все названные выше элементы и не содержит биологических ошибок | 3 |
| Ответ включает 2 из названных выше элементов и не содержит биологических ошибок,
ИЛИ ответ включает все названные выше элементы, но содержит негрубые биологические ошибки |
2 |
| Ответ включает 1 из названных выше элементов и не содержит биологических ошибок,
ИЛИ ответ включает 2 из названных выше элементов, но содержит негрубые биологические ошибки |
1 |
| Ответ неправильный | 0 |
| Максимальный балл | 3 |
Раздел: Основы генетики
В настоящее время важное место в сфере медицинских наук занимает медицинская генетика, которая изучает генетические заболевания. Вы уже знаете, что
вариантов мутаций множество: от выпадения отдельных нуклеотидов в гене до утраты целых хромосом. Количество вариантов мутаций и их сочетаний — бесчисленно, что делает медицинскую генетику неисчерпаемой.
Медицинская генетика играет важную роль при планировании семьи, служит для предупреждения наследственных заболеваний. В данной статье мы изучим некоторые наиболее известные наследственные заболевания.

Альбинизм
Альбинизм (лат. albus — белый) — врожденное заболевание, наследуемое по рецессивному типу и связанное с нарушением синтеза
черного пигмента — меланина (греч. melanos – черный) у животных или хлорофилла (у растений). Альбинизм возникает в результате
генной мутации в участке ДНК, ответственном за синтез меланина/хлорофилла.
Растения с утратой хлорофилла утрачивают способность улавливать солнечный свет, поэтому полный альбинизм для них
заканчивается летально. У животных мутация происходит в гене тирозиназы, в связи с чем меланин не синтезируется: кожа
альбиносов не способна загорать, для них характерен больший риск ожогов и рака кожи.
Радужка пропускает свет и становится красноватого оттенка, за счет кровеносных сосудов, расположенных на глазном дне.

Серповидно-клеточная анемия
Это наследственное заболевание, вызванное генной мутацией, в результате которой меняется конформация молекулы гемоглобина:
эритроцит становится выгнутым и напоминает серп.
Эта болезнь встречается особенно часто в странах, эндемичных по малярии. Больные серповидно-клеточной анемией обладают
повышенной устойчивостью к заражению малярийным плазмодием, поэтому эту болезнь можно рассматривать как результат действия
естественного отбора: с ней выживаемость людей повышалась, и они продолжали род, передавая мутацию потомкам.

Синдром Дауна
Наследственное заболевание, возникающее в результате геномной патологии: трисомия по 21-ой паре хромосом. Это означает, что
вместо двух хромосом в 21-ой паре появляется одна лишняя — третья хромосома. Причина ее появления связана с нерасхождением
хромосом во время мейоза.
Риск рождения ребенка с синдромом Дауна возрастает с увеличением возраста матери.

Синдром проявляется характерными признаками: плоское лицо, приоткрытый рот, поперечная ладонная складка, гиперподвижность суставов,
эпикантус (кожная складка, прикрывающая угол глазной щели).

Синдром Эдвардса
Наследственное заболевание, вызванное геномной мутацией — трисомией по 18 паре хромосом. Причина — нерасхождение хромосом во время
мейоза, еще до оплодотворения. Чаще болезнь встречается у пожилых матерей.
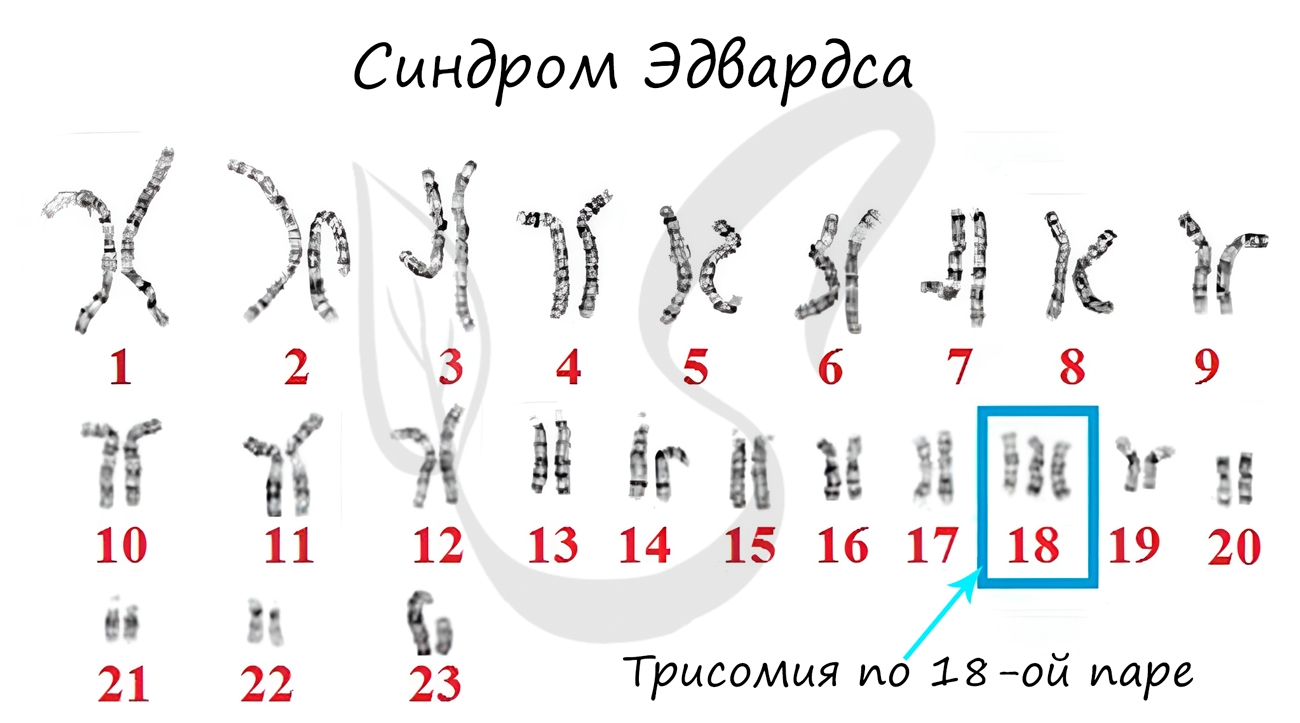
Детям с синдромом Эдвардса сопутствуют пороки сердца и сосудов: 60% детей умирают в течение первых 3 месяцев, до 1 года доживают лишь 5-10% детей.
Синдром Патау
Наследственное заболевание, обусловленной геномной мутацией — трисомией по 13 паре хромосом. Существует зависимость между возрастом матери
и вероятностью рождения ребенка с синдромом Патау (с возрастом риск увеличивается), хотя зависимость менее выражена, чем в случае с синдромом
Дауна.
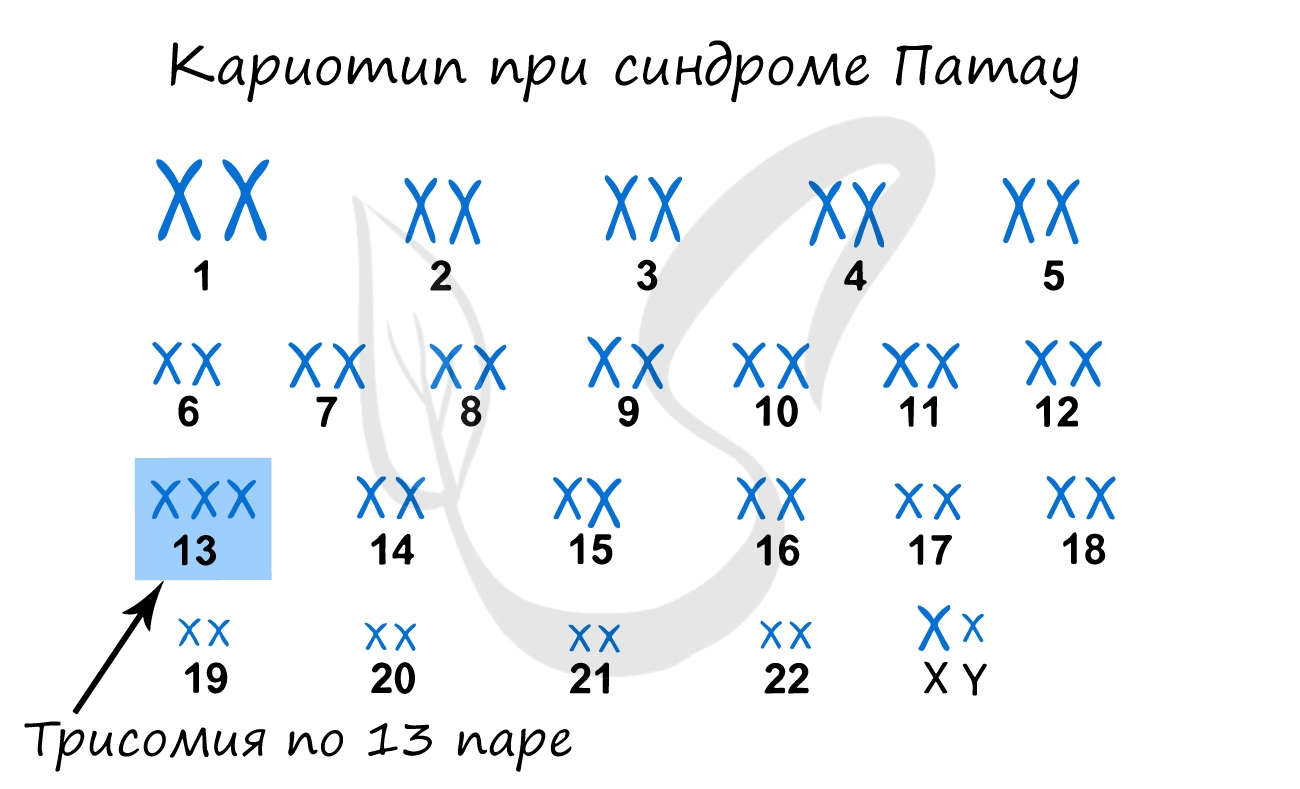
При данном синдроме обнаруживаются тяжелые врожденные пороки сердца и сосудов, нервной системы. Большинство детей с синдромом Патау умирают
в первые недели или месяцы жизни, до 1 года доживают лишь 5% детей.

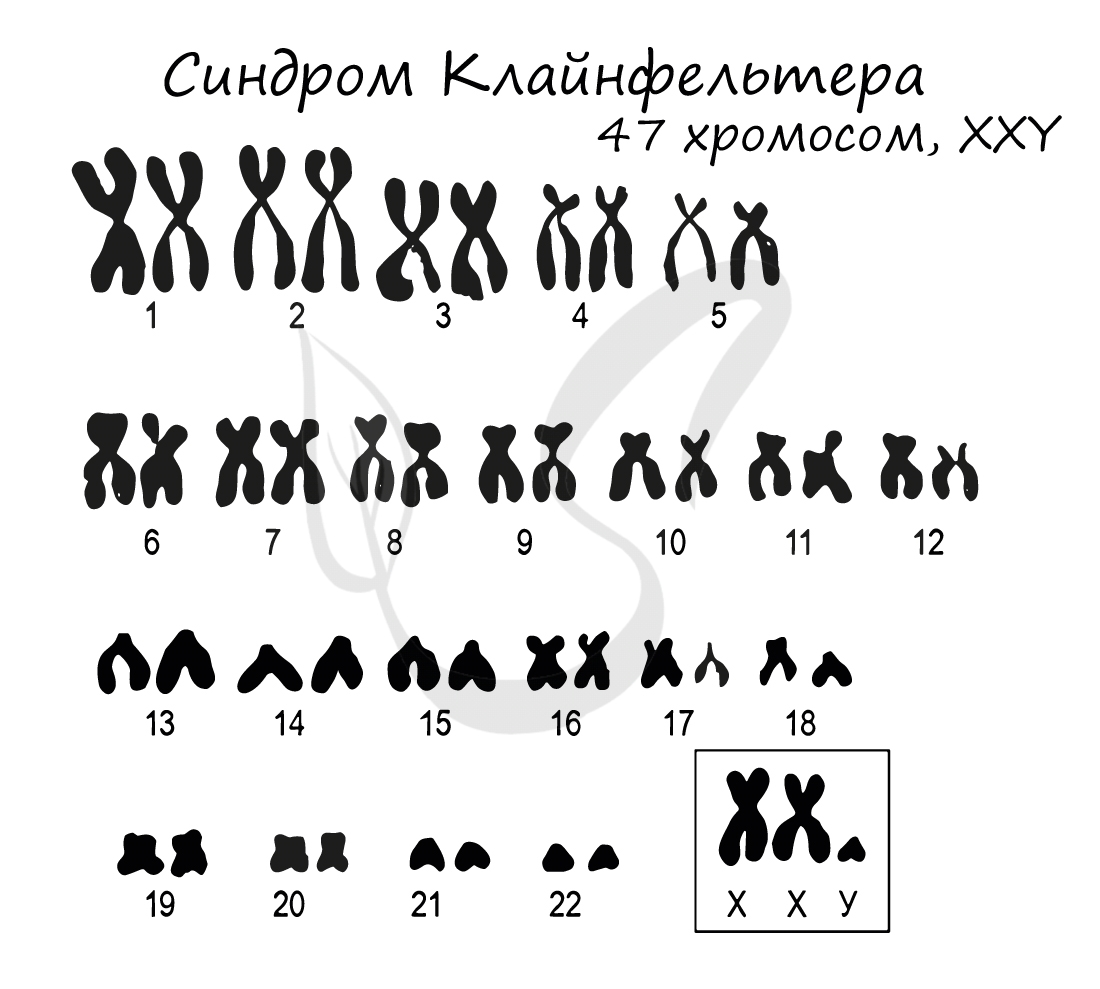
Синдром Клайнфельтера
Синдром Клайнфельтера представляет собой наследственное заболевание, развивающееся вследствие полисомии по X и Y хромосомам (половым
хромосомам). Возможны несколько вариантов генотипов: XXY (самый частый), XYY, XXXY, XXXXY, XXXYY. На всякий случай напомню норму
мужского генотипа — «XY» 
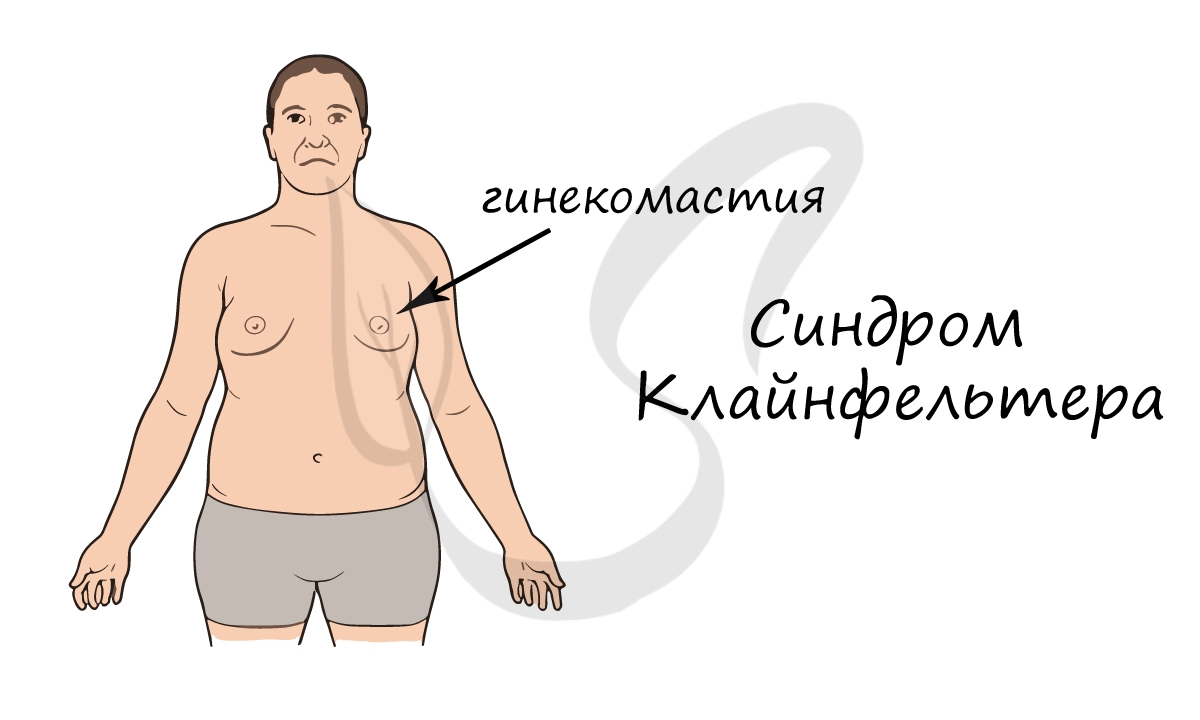
Диагностируется синдром относительно поздно, так как проявляется только после полового созревания. В подростковом возрасте развивается гинекомастия
(увеличение грудной железы), сохраняющаяся всю жизнь. Характерно наличие маленьких плотных яичек. Синдром Клайнфельтера приводит к бесплодию.
Синдром Шерешевского-Тернера
Синдром Шерешевского-Тернера — наследственное заболевание, характерное только для женщин и возникающее в результате моносомии по половым
хромосомам. Генотип человека при таком заболевании будет записан как X0 (45 хромосом).
Больные синдромом Шерешевского-Тернера низкорослые, инфантильные, их психический статус характеризуется состоянием беспричинно приподнятого
настроения — эйфорией. Тем не менее, интеллект и жизнеспособность сохранены. Из-за геномной мутации (X0) стерильны.

© Беллевич Юрий Сергеевич 2018-2023
Данная статья написана Беллевичем Юрием Сергеевичем и является его интеллектуальной собственностью. Копирование, распространение
(в том числе путем копирования на другие сайты и ресурсы в Интернете) или любое иное использование информации и объектов
без предварительного согласия правообладателя преследуется по закону. Для получения материалов статьи и разрешения их использования,
обратитесь, пожалуйста, к Беллевичу Юрию.
- Главная
- Анализы
- Справочник заболеваний
- Синдром Клайнфельтера
Синдром Клайнфельтера
![]()
Григорьев Георгий Константинович
Уролог, Кандидат медицинских наук,
Врач ультразвуковой диагностики
Как выявить синдром Клайнфельтера
Синдром Клайнфельтера — это хромосомная патология, которая обусловлена появлением в кариотипе, характерном для мужского пола дополнительных Х хромосом. Частота встречаемости не превышает 1 случая на 1000 мальчиков.
Кариотип
 Наибольший процент цитогенетических типов отводится 47 XXY, реже можно выявить появление мозаицизма 46XY/47XXY, либо 46 XX/47XXY.
Наибольший процент цитогенетических типов отводится 47 XXY, реже можно выявить появление мозаицизма 46XY/47XXY, либо 46 XX/47XXY.
Крайне редко в клинической практике можно встретить полисомию 48XXXY, 48XXYY, или 49 XXXXY.
В случае мозаичного варианта отмечается сохранение части клеток с нормальным кариотипом, при котором у мужчин будут нормально развиваться половые органы с полноценным функционированием и репродуктивной способностью.
Причины развития синдрома Клайнфельтера
В основе развития синдрома Клайнфельтера лежит хромосомная аберрация, при которой хромосомы не расходятся в процессе мейоза или нарушается процесс деления зиготы. Чаще получение подобной хромосомы встречается от материнского организма. Способствовать подобным нарушениям может:
- Вирусная инфекция.
- Поздняя беременность.
- Нарушение работы иммунной системы.
Симптомы и первые признаки
Дети, у которых диагностируют синдром Клайнфельтера при рождении не будут отличаться по массо-ростовым показателям, имеют правильную дифференцировку и размеры половых органов. При этом задерживается моторное и речевое развитие.
По мере взросления, к 5-8 годам рост превышает нормативные показатели, а телосложение становится диспропорциональным.
К характерным признакам синдрома Клайнфельтера относят:
- Позднее формирование вторичных половых признаков.
- Гипоплазию яичек.
- Малые размеры полового члена.
- Гинекомастию.
- Брадикардию, акроцианоз, потливость стоп.
- Сниженное половое влечение.
- Бесплодие.
Диагностика синдрома Клайнфельтера
Подтвердить диагноз синдрома Клайнфельтера можно ещё до рождения ребёнка. Для этого выполняется инвазивные пренатальные процедуры, например:
 Амниоцентез.
Амниоцентез.- Кордоцентез.
 Амниоцентез.
Амниоцентез.В постнатальном периоде заподозрить наличие диагноза может эндокринолог, андролог или генетик. Помимо внешнего осмотра, выяснения жалоб и данных анамнеза, а также репродуктивной функции, назначают:
- Исследование полового хроматина со слизистой щёк для выявления телец Барра.
- Определение кариотипа.
- Ультразвуковое исследование органов мошонки. Необходимо помнить, что отсутствие яичек в мошонке может быть косвенным признаком заболевания.
- Спермограмму.
- Оценку гормонального профиля.
- Биопсию яичек.
Лечение
Из-за невозможности вмешаться в гормональный фон пациента, добиться полного излечения невозможно.
Терапия при синдроме Клайнфельтера носит симптоматический характер, включая:
- Приём гормональных средств.
- Антидепрессанты или другие психотерапевтические препараты.
Для улучшения речевого развития показаны занятия с логопедом, а с целью предупреждения частых заболеваний — закаливание и ЛФК.
Чтобы повысить трудоспособность и социальную адаптацию рекомендуются психотерапевтические мероприятия.
Впервые о синдроме Клайнфельтера заговорили еще в 40-х годах ХХ века. Сейчас это генетическое нарушение подробно изучается. По разным оценкам, оно встречается у одного из 450-700 новорожденных мальчиков. Зарубежные врачи называют синдром Клайнфельтера третьим по распространенности среди всех эндокринных заболеваний. Чаще него встречаются только сахарный диабет и патологии щитовидной железы. И при этом до сих пор заболевание остается сложно диагностируемым — только у 10% больных синдром выявляется в детстве. MedAboutMe расскажет, какие методы диагностики и лечения внедряются в современной медицине.
Синдром Клайнфельтера и его симптомы

Синдром Клайнфельтера — хромосомная патология, которая бывает только у мальчиков. У здорового человека 23 пары хромосом, последняя из которых отвечает за пол: XY — мужчина, XX — женщина. При наиболее распространенном варианте заболевания у мальчика вместо нормального генотипа обнаруживается лишняя женская хромосома. Поэтому часто болезнь называют XXY-синдромом. Однако сегодня известны и другие формы хромосомных нарушений:
- 47 хромосом — XYY.
- 48 хромосом — XXXY, XYYY, XXYY
- 49 хромосом — XXXXY, XXXYY.
Чем больше лишних хромосом, тем более выражены симптомы заболевания. Однако у детей с распространенным вариантом XXY заподозрить синдром Клайнфельтера бывает очень трудно. Выраженные признаки болезни, по сути, проявляются уже во взрослом возрасте, когда у мужчины диагностируется бесплодие.
Среди симптомов в период полового созревания могут проявляться следующие:
- Высокий рост.
- Диспропорциональность конечностей.
- Феминное телосложение, с широкими бедрами и узкими плечами.
- Гинекомастия (увеличение молочных желез).
- Недоразвитость половых органов.
- Задержка умственного развития разной степени или умственная отсталость. Наблюдается у 50-60% больных.
- Аномалии костной ткани.
Диагностика заболевания: тесты и методики

Синдром Клайнфельтера диагностируется просто — достаточно провести кариотипирование, которое точно подтвердит или опровергнет диагноз. Однако основная проблема в том, что такие тесты назначаются крайне редко и никак не в числе скрининговых обследований. Поэтому люди с типом XXY и невыраженными симптомами заболевания могут всю жизнь даже не подозревать о наличии синдрома. При этом с возрастом будут проявляться следующие проблемы:
- Бесплодие.
- Эректильная дисфункция.
- Остеопороз.
- Болезни сердца (по некоторым данным, хромосомное нарушение повышает риск развития инфаркта).
- Рак молочной железы (увеличение грудных желез дает предпосылки для развития этой болезни у мужчин).
Поскольку синдром широко распространен, врачи работают над тем, чтобы найти эффективные методы диагностики. Трудность в том, что это генетическое заболевание не может передаваться по наследству — мужчины, страдающие синдромом, бесплодны. Другие однозначные факторы риска на сегодняшний день не выявлены, как и причины возникновения патологии.
Обнаружить нарушение можно еще в пренатальном периоде. Для этого ранее предлагалось два вида диагностики:
- Биопсия хориона (делается на 9-12-й неделе беременности).
- Амниоцентез (делается на 16-18-й неделе беременности).
Оба обследования очень точны, но предполагают хирургическое вмешательство. И хотя риск серьезных осложнений (выкидыши встречаются у 1%) невелик, женщины часто отказываются от диагностики. Мотивируют такое решение тем, что конкретных показаний для проведения обследования нет.
Начиная с 2012 года, проблема пренатальной диагностики была частично решена — ученые США разработали ДНК-тест Panorama. Для обследования достаточно взять кровь из вены женщины. Такие неинвазивные тесты выявляют целый ряд патологий, в том числе и синдром Клайнфельтера. Проводить анализ можно начиная с 9-й недели беременности.
В 2015 году специалисты Медицинского центра Колумбийского университета (CUMC) начали разработку метода диагностики, который поможет заподозрить синдром в детском возрасте. «Индекс физического фенотипа Клайнфельтера» (KSPHI) сегодня включает 13 признаков болезни. Так, педиатры должны обратить внимание на рост, размах рук, окружность талии, развитие молочных желез, недоразвитость семенников и прочее. «Этот новый инструмент оценки поможет врачам выявлять потенциальных больных уже при первичной медико-санитарной помощи», — говорит соавтор исследования доктор Елена Фенной. Диагностика заболевания в детстве крайне важна, ведь чем раньше начнется лечение, тем менее выраженными будут осложнения хромосомной патологии.
Гормоны и другие методы лечения

Вылечить синдром Клайнфельтера невозможно, однако люди с таким диагнозом могут жить полноценной жизнью. Основная проблема, с которой сталкиваются такие пациенты — нехватка мужских половых гормонов. Именно это приводит к феминизации и проявлению вторичных женских половых признаков. У мальчиков увеличивается грудь, наблюдаются жировые отложения в зоне бедер, не грубеет голос.
Одним из распространенных способов лечения патологии является гормонотерапия. Мужские половые гормоны (тестостерон) назначаются пациентам пожизненно. При этом врачи отмечают, что это лишь часть реабилитации больных синдромом Клайнфельтера. Специалисты Медицинского центра Колумбийского университета (CUMC), которые разрабатывают методику диагностики болезни, подробно изучали истории болезней пациентов с этой патологией. По их данным, у 38% участников с синдромом Клайнфельтера была низкая самооценка, у 26% — плохая самооценка, а у 16% — депрессии. Примечательно, что это показатели взрослых пациентов, получающих замещающую гормонотерапию. «Данные свидетельствуют о том, что лечение тестостероном не может устранить те психоэмоциональные проблемы, с которыми сталкиваются люди с синдромом Клайнфельтера», — отмечает соавтор исследования Елена Фенной.
Чем позже начато лечение, тем менее эффективным оно будет. Например, если синдром диагностирован в детском возрасте, терапия, направленная на профилактику гинекомастии, может полностью предупредить развитие этого отклонения. Взрослые же пациенты должны проходить операцию по удалению молочных желез (мастэктомию).
Без должного лечения изменения внешности отражаются на жизни мальчика и подростка, сильно затрудняют социализацию. Поэтому пациенты, у которых синдром диагностирован во взрослом возрасте, должны наряду с другим лечением посещать психотерапевта.
Люди с синдромом Клайнфельтера, в генотипе которых больше хромосом Х, могут делать операции по смене пола. В этом случае они пожизненно должны принимать женские гормоны.
Борьба с бесплодием

Главная проблема мужчин, страдающих синдромом Клайнфельтера — бесплодие. Вовремя начатая гормонотерапия поможет вести полноценную половую жизни и избежать эректильной дисфункции, но не способна восстановить фертильность.
До недавнего времени пациенты с XXY синдромом считались полностью бесплодными. Однако для людей с таким диагнозом выходом могут стать новые технологии экстракорпорального оплодотворения (ЭКО). У мужчин с синдромом Клайнфельтера наблюдаются разные показатели спермограмм — есть пациенты, у которых вообще нет сперматозоидов, но есть и те, у которых они присутствуют в малом количестве. Для вторых успешно используется вспомогательный метод ИКСИ (интрацитоплазматическая инъекция сперматозоида), при котором сперматозоид вводится в яйцеклетку с помощью иглы. По медицинским данным, это дает возможность почти четверти мужчин с синдромом Клайнфельтера стать биологическими отцами.
Даже пациентам с азооспермией, полным отсутствием сперматозоидов в эякуляте, врачи сегодня дают шанс стать родителями. Новые исследования говорят о том, что зародышевые клетки могут находиться непосредственно в яичках больного. И в этом случае для получения сперматозоида проводится биопсия органа.
Также врачи предполагают, что разрушение яичка и последующие проблемы с фертильностью и эрекцией возникают в подростковом возрасте, в период полового созревания. Если же синдром диагностирован раньше, есть шанс взять сперму или ткань яичка и сохранить их с помощью криоконсервации до начала патологических изменений.
Возможность получения сперматозоидов у мужчин с немозаичной формой синдрома Клайнфельтера в программах экстракорпорального оплодотворения. Обзор литературы и описание случая / Витязева И. И., Боголюбов С.В., Брагина Е.Е. // Андрология и генитальная хирургия – 2014 – 3
Атипичные формы синдрома Шерешевского-Тернера и Клайнфельтера / АТИПИЧНЫЕ ФОРМЫ СИНДРОМА ШЕРЕШЕВСКОГО // Медицинская генетика – 2005 – Т. 4 №6
Синдром Клайнфельтера — особенности заболевания

Синдром Клайнфельтера — это генетическое заболевание, связанное с аномальным количеством половых хромосом. 0,1-0,2% мальчиков рождаются с этим синдромом. Мужчины с этим синдромом очень часто борются с многочисленными проблемами со здоровьем, особенно с бесплодием.
Что такое синдром Клайнфельтера?
В правильно развивающихся клетках плода насчитывается 23 пары хромосом. Однако бывает, что на ранних сроках беременности происходит ненормальное деление клеток, и вместо 46 хромосом их будет больше или меньше. Такая ситуация возникает при синдроме Клайнфельтера, когда XXY присутствует в паре половых хромосом вместо XX или XY. В этом случае родится мальчик, но с определенным набором дефектов и нарушений здоровья.
В связи с половыми хромосомами мы также можем иметь дело с синдромом Тернера (моносомия, X) или синдромом гиперженщины (трисомия, XXX).
Как распознать синдром Клайнфельтера при беременности?
Пренатальное тестирование — это простой и эффективный способ обнаружить синдром Клайнфельтера в утробе матери. Благодаря этому можно рано подготовиться к воспитанию ребенка, у которого будут какие-то проблемы. Раннее знание заболевания также важно с клинической точки зрения, так как оно позволит врачу узнать о причинах заболевания и возможных методах лечения.
Генетические пренатальные тесты проводятся после 10 недель беременности. Тогда можно получить достаточное количество ДНК ребенка из крови матери, чтобы проверить его хромосомы. Для этого проводятся, например, тесты:
- Тест NIFTY Pro;
- Sanco test — тест на весь геном;
- Тест Sanco Plus;
- Тест на гармонию.

Это неинвазивные тесты, так как для них требуется только образец венозной крови от матери. Сфера их применения включает анеуплоидию половых хромосом, трисомы (включая синдромы Дауна и Эдвардса ) и некоторые синдромы делеции и дупликации (например, синдром кошачьего крика).
Синдром Клайнфельтера — характерные симптомы
Синдром Клайнфельтера редко диагностируется в детском возрасте, поскольку специфические симптомы наиболее выражены только в период полового созревания. Изначально, в детстве, у пациента может быть только меньший объем яичек, недоразвитие полового члена, а также незначительные нарушения физического развития.
Такой ребенок, прежде всего, растет быстрее, чем в среднем для своего возраста, но даже в дошкольном и младшем школьном возрасте у него может быть повышенное количество жировой ткани и метаболический синдром.
Позже половое созревание значительно замедляется. Может развиться гинекомастия. Яички и половой член все еще остаются маленькими. Однако у взрослых наблюдается плохое либидо и эректильная дисфункция.

Синдром Клайнфельтера связан не только с физическими, но и с психосоциальными симптомами. Нарушение речевого развития. Рано замечают трудности с обучением и концентрацией. У мужчин с этим синдромом проблемы с социальным функционированием. Кроме того, они часто более эмоциональны, чем другие мужчины.
Проблемы со здоровьем при синдроме Клайнфельтера
Синдром Клайнфельтера — наиболее частая генетическая причина бесплодия из-за первичной недостаточности яичек, размер которых меньше среднего. Более того, сперма содержится в сперме только у 8% мужчин с этим синдромом. Таким образом, отцовство возможно практически во всех случаях только при медицинской поддержке оплодотворения.
Мужчины с синдромом Клайнфельтера имеют повышенный риск развития:
- Рака молочной железы;
- Метаболического синдрома, то есть диабета, гипертонии, ожирения и нарушения липидного обмена;
- Резистентности к инсулину;
- Сердечно-сосудистых заболеваний;
- Варикозного расширения вен;
- Остеопороз;
- Тромбоз;
- Аутоиммунные заболевания.
Поэтому людей с этим синдромом следует регулярно обследовать. Прежде всего, рекомендуется анализ крови, ферментов печени, уровня ПСА и минеральной плотности костей.
Ошибка: Контактная форма не найдена.
Клиника абортов и контрацепции в Санкт-Петербурге — отделение медицинского гинекологического объединения «Диана»
Запишитесь на прием, анализы или УЗИ через контактную форму или по т. +8 (812) 62-962-77. Мы работаем без выходных с 09:00 до 21:00.
Мы находимся в Красногвардейском районе, рядом со станциями метро «Новочеркасская», «Площадь Александра Невского» и «Ладожская».
Стоимость медикаментозного аборта в нашей клинике 3300 руб. В стоимость входят все таблетки, осмотр гинеколога и УЗИ для определения сроков беременности.
Testicular Disorders
Shlomo Melmed MB ChB, MACP, in Williams Textbook of Endocrinology, 2020
Klinefelter Syndrome
Classically, Klinefelter syndrome is characterized by very small, firm testes; azoospermia and infertility; varying degrees of androgen deficiency and eunuchoidism; and uniformly elevated gonadotropin concentrations.20,221,222 It is the most common sex chromosome abnormality and the most common cause of primary hypogonadism, resulting in androgen deficiency and impaired sperm production. It occurs prenatally and neonatally in 1 of every 500 to 700 males, and the prevalence in adults is 1 in 2500.1,2 Because the syndrome does not cause premature death in boys, the low adult prevalence indicates that Klinefelter syndrome is often overlooked and underdiagnosed in men. Given the almost uniform finding of extremely small testes and other phenotypic abnormalities, it is surprising that about 75% of men with Klinefelter syndrome are never diagnosed. This might be due to failure to recognize mild phenotypes or failure to perform a testicular exam. The risk of having a child with Klinefelter syndrome increases with both maternal and paternal age.
The chromosomal abnormality in Klinefelter syndrome is the presence of one or more extra X chromosomes due to maternal meiotic nondisjunction (mostly in meiosis I) in approximately 50% of the cases or paternal meiotic nondisjunction in the remaining cases.20,221,222 The principal karyotype in 90% of men with Klinefelter syndrome is 47,XXY. Most of the remaining 10% have mosaic Klinefelter syndrome (47,XXY/46,XY), in which there is a 47,XXY karyotype in some tissues or cells and a normal 46,XY karyotype in other tissues or cells. Mosaicism occurs as a result of postfertilization mitotic nondisjunction. Men with mosaic Klinefelter syndrome usually demonstrate a variable and less severe phenotype that depends on the specific tissues in which an extra X chromosome is present. Some men with mosaicism have a normal karyotype in the testis with intact spermatogenesis and fertility. Rarely, men with Klinefelter syndrome have morethan one extra X chromosome (e.g., 48,XXXY, 49,XXXXY). Men with these variants manifest a more severe phenotype than is seen in classic Klinefelter syndrome.
Infants with Klinefelter syndrome may manifest micropenis, hypospadias, cryptorchidism, or developmental delay.222,223 During childhood, boys with the syndrome commonly have small testes and reduced penile length relative to age-matched normal individuals and may manifest relatively tall stature, clinodactyly, hypertelorism, gynecomastia, elbow dysplasia, high-arched palate, hypotonia, language delay or learning and reading disabilities requiring therapy, and behavioral problems.222,224 However, these manifestations may be mild and are often missed. Fewer than 10% of boys with Klinefelter syndrome (usually those with the most severe phenotype) are diagnosed before puberty. At puberty, the testes fail to increase in size and become firm due to a progressive loss of germ cells and seminiferous tubule hyalinization and fibrosis; Sertoli cell products, inhibin B, and AMH decline to very low or undetectable serum concentrations; circulating testosterone concentrations increase but to less than normal concentrations in some boys, resulting in varying degrees of eunuchoidism and gynecomastia; and FSH concentrations are disproportionately elevated relative to LH concentrations. Elevation of serum LH disproportionately to FSH is an indication of a disorder other than Klinefelter syndrome or any cause of primary hypogonadism that affects both Sertoli cell and Leydig cell dysfunction—for example, rare diseases such as disorders of testosterone biosynthesis or abnormalities of LH or its receptor should be considered.
Klinefelter Syndrome
Shanlee M. Davis, Judith L. Ross, in Encyclopedia of Endocrine Diseases (Second Edition), 2019
Abstract
Klinefelter syndrome (KS) is a genetic condition resulting from an additional X chromosome in phenotypic males (47,XXY). KS affects ~ 1 in 600 males however historically only ~ 25% are accurately diagnosed in their lifetime. Testicular development and function in impaired, often resulting in hypergonadotropic hypogonadism and the classic physical phenotype of small testes, long extremities, gynecomastia, and azoospermia. Testosterone replacement is standard treatment, however there is practice variability on when to initiate this therapy. Other frequently associated manifestations include mild developmental delays, language-based learning disabilities and disorders of insulin resistance which add to the increased morbidity and mortality in this condition. There are multiple mechanisms proposed for how the additional X chromosome results in the physical phenotype of KS and the heterogeneity among affected individuals. Recent advances in KS include early diagnosis identified through noninvasive prenatal screening and successful retrieval of viable sperm through testicular sperm extraction.
Read full chapter
URL:
https://www.sciencedirect.com/science/article/pii/B9780128012383661378
Disorders of Sex Development
Shlomo Melmed MB ChB, MACP, in Williams Textbook of Endocrinology, 2020
Klinefelter Syndrome and Its Variants
Klinefelter syndrome and its variants are the most common forms of sex chromosome aneuploidy, with a reported incidence of 1 in 500 to 1 in 1000 live male births.180 This incidence may be increasing.181 The classic form of Klinefelter syndrome is associated with a 47,XXY karyotype and is caused by meiotic nondisjunction of the sex chromosomes during gametogenesis (Fig. 24.18).18 This abnormality occurs during spermatogenesis in approximately 50% of patients and during oogenesis or postzygotic division in approximately 50%.180 Mosaic forms of Klinefelter syndrome (46,XY/47,XXY) likely represent mitotic nondisjunction within the developing zygote and are thought to occur in approximately 10% of individuals with Klinefelter syndrome. Other chromosomal variants of Klinefelter syndrome (e.g., 48,XXXY) can be seen.
The clinical features of Klinefelter syndrome and its variants are summarized inTable 24.4.180 In the most obvious situations, a young man may be diagnosed because of small testes, gynecomastia, poor androgenization at puberty, eunuchoid proportions,or infertility. Other features, such as learning difficulties, speech and language delay, behavioral issues, and altered motor development, may occur, and early educational support focusing on any specific areas of difficulty is important.182,183 It is likely that the clinical detection of Klinefelter syndrome based on postnatal karyotyping is biased toward detection of those individuals with a more severe phenotype, and it is estimated that as few as 25% of men with Klinefelter syndrome may be diagnosed throughout their life span. However, this pattern may change as diagnosis of Klinefelter syndrome on prenatal genetic testing becomes increasingly common.
The development of testes and a male phenotype in individuals with Klinefelter syndrome provides important evidence for the key role of the presence of the Y chromosome (rather than X chromosome number) in testis determination and subsequent prenatal androgen production. However, testicular function may not be completely intact; micropenis and hypospadias may be presenting features in some cases, and some studies report mildly elevated FSH concentrations during the postnatal “minipuberty” together with testosterone levels in the lower half of the normal range.184
A more predictable elevation in gonadotropin concentrations (e.g., FSH, LH) occurs in the periadolescent period in patients with Klinefelter syndrome, after activation of the HPG axis.185,186 By midadolescence, plasma concentrations of FSH are increased in 90% of patients with Klinefelter syndrome, and plasma concentrations of LH are increased in 80%. Other serum markers of testicular function (e.g., peripubertal inhibin B, midpubertal INSL3) are often below the normal ranges.173,186 Although puberty usually starts at a normal age with an appropriate rise in testosterone in classic Klinefelter syndrome, serum testosterone levels off around midpuberty with an increase of gonadotropins. In the majority of young adults, serum testosterone is in the lower half of the reference range.185,187 Testes usually remain small and firm; the median length and volume are 2.5 cm and 3 mL, but most are smaller than 3.5 cm (range 1–7 mL).185 Testes typically appear inappropriately small for the degree of androgenization. The serum estradiol concentration is often elevated, which contributes to the gynecomastia observed during the adolescent period.
Klinefelter’s Syndrome
François Pralong, in Encyclopedia of Endocrine Diseases, 2004
Clinical Presentation
Klinefelter’s syndrome is present in about 1 in 1000 living males. Azoospermia, gynecomastia, and the presence of small atrophic testes in otherwise normal males are the clinical hallmarks of the disease. The characteristic features of patients affected with Klinefelter’s syndrome usually first become apparent during adolescence. In a classical scenario, the diagnosis is suggested by the presence of small testes (less than 3 cm in length), commonly but not always accompanied by bilateral gynecomastia, in adolescent boys seeking medical advice because of a delayed onset of puberty. These features can be associated with a variable degree of eunuchoidism, that is, altered body proportions and tall stature. However, in particular instances, the diagnosis of Klinefelter’s syndrome can also be made as early as during the neonatal period or as late as during adult life.
Klinefelter’s syndrome can be diagnosed during the first year of life in children evaluated for microphallus, hypospadias, or cryptorchidism. Later during childhood but before adolescence, the diagnosis may be suspected on the basis of a subnormal testicular size. Indeed, in normal men, more than 80% of the total testicular volume is accounted for by the seminiferous tubules, the anatomical structures where spermatogenesis takes place. In Klinefelter’s syndrome, an abnormal fibrosis of these tubules begins before puberty. When this pathological process is severe enough, atrophy of the testes may already become evident during mid-childhood. At the other end of the spectrum of clinical presentations, a number of patients can achieve significant virilization and go undiagnosed until adulthood. These patients can be discovered incidentally during the workup of a malignancy but are usually diagnosed on the occasion of their evaluation for infertility.
Serum levels of the male hormone testosterone are normally low in prepubertal boys and begin to rise during early puberty. In boys affected with Klinefelter’s syndrome, a decreased capacity of the testes to secrete testosterone becomes evident in many cases as puberty continues. Therefore, testosterone levels tend to be either low-normal or frankly decreased in adult patients. This occurs despite an elevation above normal values of the circulating levels of luteinizing hormone (LH), the hormone that normally stimulates testosterone production by the testes. Because of the lack of testosterone, affected patients rarely become normally virilized. On the other hand, the levels of the female hormone estradiol, which is also normally secreted by the human testes, are usually elevated in Klinefelter’s patients. These anomalies result in an imbalance between the two sex hormones, with increased estradiol levels and diminished testosterone values producing a hormonal milieu favorable to the development of bilateral gynecomastia. This is observed in more than 90% of Klinefelter’s patients.
Read full chapter
URL:
https://www.sciencedirect.com/science/article/pii/B0124755704007988
Disorders of Sexual Development : Etiology, Evaluation, and Medical Management
Alan W. Partin MD, PhD, in Campbell-Walsh-Wein Urology, 2021
Klinefelter Syndrome and Variants
In 1942, Klinefelter, Reifenstein, and Albright described a syndrome characterized by eunuchoidism, gynecomastia, azoospermia, increased gonadotropin levels, and small, firm testes. By 1959, these patients were noted to have a 47,XXY karyotype (Jacobs and Strong, 1959).
Klinefelter syndrome represents the most common major abnormality of sexual development. By definition, males with at least one Y chromosome and at least two X chromosomes have Klinefelter syndrome. The classic 47,XXY complement arises as a result of nondisjunction during meiosis; it occurs in 1 of 600 liveborn males (Morris et al, 2008). But the phenotype is also associated with 48,XXYY and 49,XXXYY, and an exaggerated form of the phenotype is associated with 48,XXXY and 49,XXXXY. The mosaic form 46,XY/47,XXY is associated with milder phenotypic features of classic 47,XXY Klinefelter syndrome. Perhaps as a result of phenotypic variability, Klinefelter syndrome is believed to be underdiagnosed, with a 25% diagnostic rate in one Danish study (Bojesen et al, 2003).
In 47,XXY adults, seminiferous tubules degenerate and are replaced with hyaline. As a result, testes are firm and small, less than 3.5 cm in length. Histologically, Leydig cells appear to predominate. They are seen in large clumps in certain areas of the testes, sometimes resembling Leydig cell tumors. However, the absolute volume of Leydig cells is not increased and is probably lower than normal. Serum levels of testosterone are low-normal and those of gonadotropins are elevated. Plasma estradiol levels tend to be high, with gynecomastia the result of an increased ratio of estradiol to testosterone. The vast majority of patients are azoospermic, and the presence of sperm suggests 46,XY/47,XXY mosaicism. There does appear to be a depletion of germ cells with the onset of puberty (Wikström et al., 2004). Fertility, with the benefit of testicular sperm extraction and intracytoplasmic sperm injection (ICSI), has been reported in patients with Klinefelter syndrome. A recent meta-analysis examining ICSI outcomes reported a 43% live birth rate per ICSI cycle (Corona et al., 2017). Some infertility experts advocate coupling intracytoplasmic sperm injection with preimplantation diagnosis given the lower rate of normal embryos from Klinefelter syndrome patients (54%) versus controls (77%) (Staessen et al, 2003). However, there have been over 200 normal live births reported using ICSI without preimplantation genetic analysis, which brings into question the utility of this testing in predicting outcomes (Ni et al., 2016).
The decreased androgen production may impair normal secondary sexual development. Muscle development may be poor, and the fat distribution is more typical of females than of males. Normal amounts of pubic and axillary hair may be present, but facial hair is sparse. Patients tend to be taller than average, mainly because of the disproportionate length of their legs, which is present even in childhood. Otherwise, few if any distinguishing features are present in the prepubertal child.
Klinefelter Syndrome
M.A. Ferguson-Smith, in Brenner’s Encyclopedia of Genetics (Second Edition), 2013
Klinefelter Syndrome Variants
Patients with Klinefelter syndrome and more than two X chromosomes have greater physical and learning disabilities associated with a number of malformations. These include microcephaly, proximal radioulnar synostosis, undescended testes, congenital heart disease, cleft palate, and short incurved fifth digit. The facies is characteristic with prognathism, epicanthus, hypertelorism, myopia, strabismus, and mid-face hypoplasia. The maximum number of sex chromatin bodies per nucleus is always one fewer than the total number of X chromosomes, indicating that X-inactivation ensures that only one X chromosome is functionally active. However, abnormal dosage of X/Y homologous loci, which normally escape X-inactivation, is thought to be responsible for the level of clinical disability associated with additional X chromosomes. Male differentiation occurs irrespective of the number of X chromosomes and this attests to the dominant male-determining effect of the SRY gene on the Y chromosome. The XXY condition has been observed in a number of other species including mouse, cat, horse, and sheep; in each case, male differentiation is apparent.
Other variants of Klinefelter syndrome are known. SRY + XX males, in whom the SRY locus has been transferred to the X chromosome by accidental recombination within the differential segments of the X and Y chromosomes, show little disability other than infertility. Those with sex chromosome mosaicism, that is, XY/XXY or XX/XXY, also tend to show less disability than XXY patients. XY/XXY patients are occasionally fertile and XX/XXY patients may rarely be found to be true hermaphrodites.
Read full chapter
URL:
https://www.sciencedirect.com/science/article/pii/B9780123749840008354
Klinefelter Syndrome
Georgianne Arnold MD, in Pediatric Clinical Advisor (Second Edition), 2007
Pearls & Considerations
Comments
- •
-
Some individuals recommend that the eponym Klinefelter syndrome be reserved only for males who exhibit the characteristic phenotype.
- •
-
Those diagnosed prenatally or for other reasons who do not experience difficulties with growth or performance should be referred to as having a 47,XXY karyotype, not Klinefelter syndrome.
Patient/Family Education
- •
-
A relatively normal childhood can be expected if the child is diagnosed prenatally.
- •
-
Any behavior or learning issues may require special evaluation and school placement.
- •
-
Sexual intercourse is normal, although some men receive testosterone supplementation.
- •
-
Infertility may be amenable to sperm manipulation (i.e., ICSI) in some men.
- •
-
Support group information can be obtained through the Klinefelter Syndrome Support Group: http://klinefeltersyndrome.org/
- •
-
The Even Exchange, a newsletter of Klinefelter Syndrome and Associates, provides useful information for parents and adult patients (P.O. Box 119, Roseville, CA 95678‐0119).
- •
-
Klinefelter Syndrome, The X‐tra Special Boy, and For Boys Only are publications available from the Genetics Clinic, Crippled Children’s Division, Oregon Health Sciences University.
Read full chapter
URL:
https://www.sciencedirect.com/science/article/pii/B9780323035064101804
Mental Retardation
Sandra Cluett Redden, … Martha Pope, in Encyclopedia of the Human Brain, 2002
VII.E Klinefelter Syndrome
Klinefelter syndrome (KS) is a sex chromosome disorder and occurs due to the presence of an extra X chromosome in males (XXY). It is estimated that 1 in 700 to 1 in 900 live male births are affected by KS. Early research linked KS with psychiatric disorders, criminal behavior, and mental retardation. These studies had methodological concerns that impacted validity; however, many still hold to these early findings. In truth, males with KS are at risk for developmental, learning, language, and behavioral problems along with psychiatric disorders, but they are not inherently criminal.
Physical manifestations of KS result in impairment of normal genital and sexual development. Although persons with KS enter puberty at the typical age, inadequate testosterone levels prevent normal pubertal progress. Hypogonadism is present with a slowed development of or lack of secondary sexual characteristics. In addition, KS leads to smaller than normal testes that often contribute to infertility. Cognitive abilities range from mild mental retardation to above-average intelligence. In general, persons with KS have deficits in verbal abilities, whereas nonverbal abilities are typically in the average range. Behaviorally, individuals with KS may be reserved, withdrawn, and immature, and they are at risk for having poor peer relationships. Self-esteem problems also have been described. In contrast, they also have been described as being easygoing, underactive, compliant, and, consequently, well liked by teachers.
Read full chapter
URL:
https://www.sciencedirect.com/science/article/pii/B0122272102002041
Sex-Chromosome Abnormalities
Claus H. Gravholt, in Emery and Rimoin’s Principles and Practice of Medical Genetics (Sixth Edition), 2013
44.4.4 Fertility
KS patients are usually considered infertile, but with the development of the testicular sperm extraction (TESE) and intracytoplasmatic sperm injection (ICSI) methodologies, it is now possible to extract viable sperm from the testes by surgical biopsy and inject it directly into an extracted ovum. Worldwide more than 60 children have been born after successful ICSI treatment in KS couples (164,176). There have been concerns about an increased risk of chromosome aberrations in the offspring because an increased number of both autosomal and sex-chromosome aneuploidy has been found in spermatozoas extracted or ejaculated from KS men (177). As a consequence, professional genetic counseling and options of prenatal diagnosis or even preimplantation genetic diagnosis should always be offered to couples seeking infertility treatment where the male is suffering from KS.
Read full chapter
URL:
https://www.sciencedirect.com/science/article/pii/B9780123838346000501
Reproductive Medicine
Yetunde Ibrahim, James Hotaling, in Encyclopedia of Reproduction (Second Edition), 2018
Implications of Non-reproductive Impairments
KS is a key risk factor for early-onset osteoporosis (Bojesen et al., 2011). The etiology is likely multifactorial but testosterone is thought to play a key role. It has been well defined that testosterone in men and estrogen in women play a vital role to bone health. Thus, in KS, testosterone deficiency can lead to decreased bone formation and resorption. Other possible mechanisms for osteoporosis in KS are abnormalities in CAG length, low levels of insulin-like factor 3 and X-chromosome inactivation (Ferlin et al., 2011; Wikstrom et al., 2006). Men with KS have also been shown to be susceptible to metabolic dysfunction including type 2 diabetes, decreased muscle, increased adiposity, decreased insulin sensitivity, decreased muscle strength and oxygen consumption capacity (Gravholt et al., 2011).
Read full chapter
URL:
https://www.sciencedirect.com/science/article/pii/B9780128012383645373
Testicular Disorders
Shlomo Melmed MB ChB, MACP, in Williams Textbook of Endocrinology, 2020
Klinefelter Syndrome
Classically, Klinefelter syndrome is characterized by very small, firm testes; azoospermia and infertility; varying degrees of androgen deficiency and eunuchoidism; and uniformly elevated gonadotropin concentrations.20,221,222 It is the most common sex chromosome abnormality and the most common cause of primary hypogonadism, resulting in androgen deficiency and impaired sperm production. It occurs prenatally and neonatally in 1 of every 500 to 700 males, and the prevalence in adults is 1 in 2500.1,2 Because the syndrome does not cause premature death in boys, the low adult prevalence indicates that Klinefelter syndrome is often overlooked and underdiagnosed in men. Given the almost uniform finding of extremely small testes and other phenotypic abnormalities, it is surprising that about 75% of men with Klinefelter syndrome are never diagnosed. This might be due to failure to recognize mild phenotypes or failure to perform a testicular exam. The risk of having a child with Klinefelter syndrome increases with both maternal and paternal age.
The chromosomal abnormality in Klinefelter syndrome is the presence of one or more extra X chromosomes due to maternal meiotic nondisjunction (mostly in meiosis I) in approximately 50% of the cases or paternal meiotic nondisjunction in the remaining cases.20,221,222 The principal karyotype in 90% of men with Klinefelter syndrome is 47,XXY. Most of the remaining 10% have mosaic Klinefelter syndrome (47,XXY/46,XY), in which there is a 47,XXY karyotype in some tissues or cells and a normal 46,XY karyotype in other tissues or cells. Mosaicism occurs as a result of postfertilization mitotic nondisjunction. Men with mosaic Klinefelter syndrome usually demonstrate a variable and less severe phenotype that depends on the specific tissues in which an extra X chromosome is present. Some men with mosaicism have a normal karyotype in the testis with intact spermatogenesis and fertility. Rarely, men with Klinefelter syndrome have morethan one extra X chromosome (e.g., 48,XXXY, 49,XXXXY). Men with these variants manifest a more severe phenotype than is seen in classic Klinefelter syndrome.
Infants with Klinefelter syndrome may manifest micropenis, hypospadias, cryptorchidism, or developmental delay.222,223 During childhood, boys with the syndrome commonly have small testes and reduced penile length relative to age-matched normal individuals and may manifest relatively tall stature, clinodactyly, hypertelorism, gynecomastia, elbow dysplasia, high-arched palate, hypotonia, language delay or learning and reading disabilities requiring therapy, and behavioral problems.222,224 However, these manifestations may be mild and are often missed. Fewer than 10% of boys with Klinefelter syndrome (usually those with the most severe phenotype) are diagnosed before puberty. At puberty, the testes fail to increase in size and become firm due to a progressive loss of germ cells and seminiferous tubule hyalinization and fibrosis; Sertoli cell products, inhibin B, and AMH decline to very low or undetectable serum concentrations; circulating testosterone concentrations increase but to less than normal concentrations in some boys, resulting in varying degrees of eunuchoidism and gynecomastia; and FSH concentrations are disproportionately elevated relative to LH concentrations. Elevation of serum LH disproportionately to FSH is an indication of a disorder other than Klinefelter syndrome or any cause of primary hypogonadism that affects both Sertoli cell and Leydig cell dysfunction—for example, rare diseases such as disorders of testosterone biosynthesis or abnormalities of LH or its receptor should be considered.
Klinefelter Syndrome
Shanlee M. Davis, Judith L. Ross, in Encyclopedia of Endocrine Diseases (Second Edition), 2019
Abstract
Klinefelter syndrome (KS) is a genetic condition resulting from an additional X chromosome in phenotypic males (47,XXY). KS affects ~ 1 in 600 males however historically only ~ 25% are accurately diagnosed in their lifetime. Testicular development and function in impaired, often resulting in hypergonadotropic hypogonadism and the classic physical phenotype of small testes, long extremities, gynecomastia, and azoospermia. Testosterone replacement is standard treatment, however there is practice variability on when to initiate this therapy. Other frequently associated manifestations include mild developmental delays, language-based learning disabilities and disorders of insulin resistance which add to the increased morbidity and mortality in this condition. There are multiple mechanisms proposed for how the additional X chromosome results in the physical phenotype of KS and the heterogeneity among affected individuals. Recent advances in KS include early diagnosis identified through noninvasive prenatal screening and successful retrieval of viable sperm through testicular sperm extraction.
Read full chapter
URL:
https://www.sciencedirect.com/science/article/pii/B9780128012383661378
Disorders of Sex Development
Shlomo Melmed MB ChB, MACP, in Williams Textbook of Endocrinology, 2020
Klinefelter Syndrome and Its Variants
Klinefelter syndrome and its variants are the most common forms of sex chromosome aneuploidy, with a reported incidence of 1 in 500 to 1 in 1000 live male births.180 This incidence may be increasing.181 The classic form of Klinefelter syndrome is associated with a 47,XXY karyotype and is caused by meiotic nondisjunction of the sex chromosomes during gametogenesis (Fig. 24.18).18 This abnormality occurs during spermatogenesis in approximately 50% of patients and during oogenesis or postzygotic division in approximately 50%.180 Mosaic forms of Klinefelter syndrome (46,XY/47,XXY) likely represent mitotic nondisjunction within the developing zygote and are thought to occur in approximately 10% of individuals with Klinefelter syndrome. Other chromosomal variants of Klinefelter syndrome (e.g., 48,XXXY) can be seen.
The clinical features of Klinefelter syndrome and its variants are summarized inTable 24.4.180 In the most obvious situations, a young man may be diagnosed because of small testes, gynecomastia, poor androgenization at puberty, eunuchoid proportions,or infertility. Other features, such as learning difficulties, speech and language delay, behavioral issues, and altered motor development, may occur, and early educational support focusing on any specific areas of difficulty is important.182,183 It is likely that the clinical detection of Klinefelter syndrome based on postnatal karyotyping is biased toward detection of those individuals with a more severe phenotype, and it is estimated that as few as 25% of men with Klinefelter syndrome may be diagnosed throughout their life span. However, this pattern may change as diagnosis of Klinefelter syndrome on prenatal genetic testing becomes increasingly common.
The development of testes and a male phenotype in individuals with Klinefelter syndrome provides important evidence for the key role of the presence of the Y chromosome (rather than X chromosome number) in testis determination and subsequent prenatal androgen production. However, testicular function may not be completely intact; micropenis and hypospadias may be presenting features in some cases, and some studies report mildly elevated FSH concentrations during the postnatal “minipuberty” together with testosterone levels in the lower half of the normal range.184
A more predictable elevation in gonadotropin concentrations (e.g., FSH, LH) occurs in the periadolescent period in patients with Klinefelter syndrome, after activation of the HPG axis.185,186 By midadolescence, plasma concentrations of FSH are increased in 90% of patients with Klinefelter syndrome, and plasma concentrations of LH are increased in 80%. Other serum markers of testicular function (e.g., peripubertal inhibin B, midpubertal INSL3) are often below the normal ranges.173,186 Although puberty usually starts at a normal age with an appropriate rise in testosterone in classic Klinefelter syndrome, serum testosterone levels off around midpuberty with an increase of gonadotropins. In the majority of young adults, serum testosterone is in the lower half of the reference range.185,187 Testes usually remain small and firm; the median length and volume are 2.5 cm and 3 mL, but most are smaller than 3.5 cm (range 1–7 mL).185 Testes typically appear inappropriately small for the degree of androgenization. The serum estradiol concentration is often elevated, which contributes to the gynecomastia observed during the adolescent period.
Klinefelter’s Syndrome
François Pralong, in Encyclopedia of Endocrine Diseases, 2004
Clinical Presentation
Klinefelter’s syndrome is present in about 1 in 1000 living males. Azoospermia, gynecomastia, and the presence of small atrophic testes in otherwise normal males are the clinical hallmarks of the disease. The characteristic features of patients affected with Klinefelter’s syndrome usually first become apparent during adolescence. In a classical scenario, the diagnosis is suggested by the presence of small testes (less than 3 cm in length), commonly but not always accompanied by bilateral gynecomastia, in adolescent boys seeking medical advice because of a delayed onset of puberty. These features can be associated with a variable degree of eunuchoidism, that is, altered body proportions and tall stature. However, in particular instances, the diagnosis of Klinefelter’s syndrome can also be made as early as during the neonatal period or as late as during adult life.
Klinefelter’s syndrome can be diagnosed during the first year of life in children evaluated for microphallus, hypospadias, or cryptorchidism. Later during childhood but before adolescence, the diagnosis may be suspected on the basis of a subnormal testicular size. Indeed, in normal men, more than 80% of the total testicular volume is accounted for by the seminiferous tubules, the anatomical structures where spermatogenesis takes place. In Klinefelter’s syndrome, an abnormal fibrosis of these tubules begins before puberty. When this pathological process is severe enough, atrophy of the testes may already become evident during mid-childhood. At the other end of the spectrum of clinical presentations, a number of patients can achieve significant virilization and go undiagnosed until adulthood. These patients can be discovered incidentally during the workup of a malignancy but are usually diagnosed on the occasion of their evaluation for infertility.
Serum levels of the male hormone testosterone are normally low in prepubertal boys and begin to rise during early puberty. In boys affected with Klinefelter’s syndrome, a decreased capacity of the testes to secrete testosterone becomes evident in many cases as puberty continues. Therefore, testosterone levels tend to be either low-normal or frankly decreased in adult patients. This occurs despite an elevation above normal values of the circulating levels of luteinizing hormone (LH), the hormone that normally stimulates testosterone production by the testes. Because of the lack of testosterone, affected patients rarely become normally virilized. On the other hand, the levels of the female hormone estradiol, which is also normally secreted by the human testes, are usually elevated in Klinefelter’s patients. These anomalies result in an imbalance between the two sex hormones, with increased estradiol levels and diminished testosterone values producing a hormonal milieu favorable to the development of bilateral gynecomastia. This is observed in more than 90% of Klinefelter’s patients.
Read full chapter
URL:
https://www.sciencedirect.com/science/article/pii/B0124755704007988
Disorders of Sexual Development : Etiology, Evaluation, and Medical Management
Alan W. Partin MD, PhD, in Campbell-Walsh-Wein Urology, 2021
Klinefelter Syndrome and Variants
In 1942, Klinefelter, Reifenstein, and Albright described a syndrome characterized by eunuchoidism, gynecomastia, azoospermia, increased gonadotropin levels, and small, firm testes. By 1959, these patients were noted to have a 47,XXY karyotype (Jacobs and Strong, 1959).
Klinefelter syndrome represents the most common major abnormality of sexual development. By definition, males with at least one Y chromosome and at least two X chromosomes have Klinefelter syndrome. The classic 47,XXY complement arises as a result of nondisjunction during meiosis; it occurs in 1 of 600 liveborn males (Morris et al, 2008). But the phenotype is also associated with 48,XXYY and 49,XXXYY, and an exaggerated form of the phenotype is associated with 48,XXXY and 49,XXXXY. The mosaic form 46,XY/47,XXY is associated with milder phenotypic features of classic 47,XXY Klinefelter syndrome. Perhaps as a result of phenotypic variability, Klinefelter syndrome is believed to be underdiagnosed, with a 25% diagnostic rate in one Danish study (Bojesen et al, 2003).
In 47,XXY adults, seminiferous tubules degenerate and are replaced with hyaline. As a result, testes are firm and small, less than 3.5 cm in length. Histologically, Leydig cells appear to predominate. They are seen in large clumps in certain areas of the testes, sometimes resembling Leydig cell tumors. However, the absolute volume of Leydig cells is not increased and is probably lower than normal. Serum levels of testosterone are low-normal and those of gonadotropins are elevated. Plasma estradiol levels tend to be high, with gynecomastia the result of an increased ratio of estradiol to testosterone. The vast majority of patients are azoospermic, and the presence of sperm suggests 46,XY/47,XXY mosaicism. There does appear to be a depletion of germ cells with the onset of puberty (Wikström et al., 2004). Fertility, with the benefit of testicular sperm extraction and intracytoplasmic sperm injection (ICSI), has been reported in patients with Klinefelter syndrome. A recent meta-analysis examining ICSI outcomes reported a 43% live birth rate per ICSI cycle (Corona et al., 2017). Some infertility experts advocate coupling intracytoplasmic sperm injection with preimplantation diagnosis given the lower rate of normal embryos from Klinefelter syndrome patients (54%) versus controls (77%) (Staessen et al, 2003). However, there have been over 200 normal live births reported using ICSI without preimplantation genetic analysis, which brings into question the utility of this testing in predicting outcomes (Ni et al., 2016).
The decreased androgen production may impair normal secondary sexual development. Muscle development may be poor, and the fat distribution is more typical of females than of males. Normal amounts of pubic and axillary hair may be present, but facial hair is sparse. Patients tend to be taller than average, mainly because of the disproportionate length of their legs, which is present even in childhood. Otherwise, few if any distinguishing features are present in the prepubertal child.
Klinefelter Syndrome
M.A. Ferguson-Smith, in Brenner’s Encyclopedia of Genetics (Second Edition), 2013
Klinefelter Syndrome Variants
Patients with Klinefelter syndrome and more than two X chromosomes have greater physical and learning disabilities associated with a number of malformations. These include microcephaly, proximal radioulnar synostosis, undescended testes, congenital heart disease, cleft palate, and short incurved fifth digit. The facies is characteristic with prognathism, epicanthus, hypertelorism, myopia, strabismus, and mid-face hypoplasia. The maximum number of sex chromatin bodies per nucleus is always one fewer than the total number of X chromosomes, indicating that X-inactivation ensures that only one X chromosome is functionally active. However, abnormal dosage of X/Y homologous loci, which normally escape X-inactivation, is thought to be responsible for the level of clinical disability associated with additional X chromosomes. Male differentiation occurs irrespective of the number of X chromosomes and this attests to the dominant male-determining effect of the SRY gene on the Y chromosome. The XXY condition has been observed in a number of other species including mouse, cat, horse, and sheep; in each case, male differentiation is apparent.
Other variants of Klinefelter syndrome are known. SRY + XX males, in whom the SRY locus has been transferred to the X chromosome by accidental recombination within the differential segments of the X and Y chromosomes, show little disability other than infertility. Those with sex chromosome mosaicism, that is, XY/XXY or XX/XXY, also tend to show less disability than XXY patients. XY/XXY patients are occasionally fertile and XX/XXY patients may rarely be found to be true hermaphrodites.
Read full chapter
URL:
https://www.sciencedirect.com/science/article/pii/B9780123749840008354
Klinefelter Syndrome
Georgianne Arnold MD, in Pediatric Clinical Advisor (Second Edition), 2007
Pearls & Considerations
Comments
- •
-
Some individuals recommend that the eponym Klinefelter syndrome be reserved only for males who exhibit the characteristic phenotype.
- •
-
Those diagnosed prenatally or for other reasons who do not experience difficulties with growth or performance should be referred to as having a 47,XXY karyotype, not Klinefelter syndrome.
Patient/Family Education
- •
-
A relatively normal childhood can be expected if the child is diagnosed prenatally.
- •
-
Any behavior or learning issues may require special evaluation and school placement.
- •
-
Sexual intercourse is normal, although some men receive testosterone supplementation.
- •
-
Infertility may be amenable to sperm manipulation (i.e., ICSI) in some men.
- •
-
Support group information can be obtained through the Klinefelter Syndrome Support Group: http://klinefeltersyndrome.org/
- •
-
The Even Exchange, a newsletter of Klinefelter Syndrome and Associates, provides useful information for parents and adult patients (P.O. Box 119, Roseville, CA 95678‐0119).
- •
-
Klinefelter Syndrome, The X‐tra Special Boy, and For Boys Only are publications available from the Genetics Clinic, Crippled Children’s Division, Oregon Health Sciences University.
Read full chapter
URL:
https://www.sciencedirect.com/science/article/pii/B9780323035064101804
Mental Retardation
Sandra Cluett Redden, … Martha Pope, in Encyclopedia of the Human Brain, 2002
VII.E Klinefelter Syndrome
Klinefelter syndrome (KS) is a sex chromosome disorder and occurs due to the presence of an extra X chromosome in males (XXY). It is estimated that 1 in 700 to 1 in 900 live male births are affected by KS. Early research linked KS with psychiatric disorders, criminal behavior, and mental retardation. These studies had methodological concerns that impacted validity; however, many still hold to these early findings. In truth, males with KS are at risk for developmental, learning, language, and behavioral problems along with psychiatric disorders, but they are not inherently criminal.
Physical manifestations of KS result in impairment of normal genital and sexual development. Although persons with KS enter puberty at the typical age, inadequate testosterone levels prevent normal pubertal progress. Hypogonadism is present with a slowed development of or lack of secondary sexual characteristics. In addition, KS leads to smaller than normal testes that often contribute to infertility. Cognitive abilities range from mild mental retardation to above-average intelligence. In general, persons with KS have deficits in verbal abilities, whereas nonverbal abilities are typically in the average range. Behaviorally, individuals with KS may be reserved, withdrawn, and immature, and they are at risk for having poor peer relationships. Self-esteem problems also have been described. In contrast, they also have been described as being easygoing, underactive, compliant, and, consequently, well liked by teachers.
Read full chapter
URL:
https://www.sciencedirect.com/science/article/pii/B0122272102002041
Sex-Chromosome Abnormalities
Claus H. Gravholt, in Emery and Rimoin’s Principles and Practice of Medical Genetics (Sixth Edition), 2013
44.4.4 Fertility
KS patients are usually considered infertile, but with the development of the testicular sperm extraction (TESE) and intracytoplasmatic sperm injection (ICSI) methodologies, it is now possible to extract viable sperm from the testes by surgical biopsy and inject it directly into an extracted ovum. Worldwide more than 60 children have been born after successful ICSI treatment in KS couples (164,176). There have been concerns about an increased risk of chromosome aberrations in the offspring because an increased number of both autosomal and sex-chromosome aneuploidy has been found in spermatozoas extracted or ejaculated from KS men (177). As a consequence, professional genetic counseling and options of prenatal diagnosis or even preimplantation genetic diagnosis should always be offered to couples seeking infertility treatment where the male is suffering from KS.
Read full chapter
URL:
https://www.sciencedirect.com/science/article/pii/B9780123838346000501
Reproductive Medicine
Yetunde Ibrahim, James Hotaling, in Encyclopedia of Reproduction (Second Edition), 2018
Implications of Non-reproductive Impairments
KS is a key risk factor for early-onset osteoporosis (Bojesen et al., 2011). The etiology is likely multifactorial but testosterone is thought to play a key role. It has been well defined that testosterone in men and estrogen in women play a vital role to bone health. Thus, in KS, testosterone deficiency can lead to decreased bone formation and resorption. Other possible mechanisms for osteoporosis in KS are abnormalities in CAG length, low levels of insulin-like factor 3 and X-chromosome inactivation (Ferlin et al., 2011; Wikstrom et al., 2006). Men with KS have also been shown to be susceptible to metabolic dysfunction including type 2 diabetes, decreased muscle, increased adiposity, decreased insulin sensitivity, decreased muscle strength and oxygen consumption capacity (Gravholt et al., 2011).
Read full chapter
URL:
https://www.sciencedirect.com/science/article/pii/B9780128012383645373